This pipeline performs ATAC Seq using Nextflow
This document describes the output produced by the pipeline. Most of the plots are taken from the MultiQC report, which summarises results at the end of the pipeline.
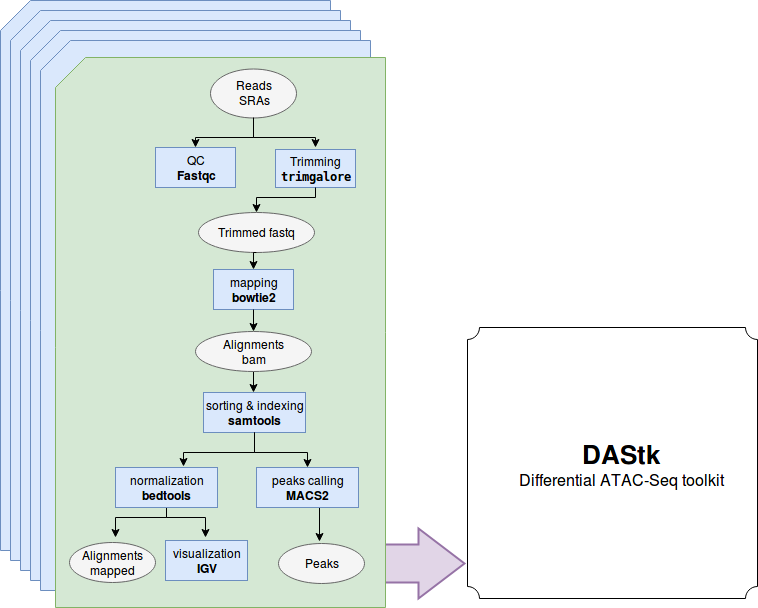
The pipeline is built using Nextflow and processes data using the following steps:
- Sra-tools --version 2.8.2 - convert sra files to fastq files
- Trim_Galore --version 0.4.4 -trimming adaptors and quality control
- FastQC --version v0.11.7 - read quality control
- MultiQC --version 1.5 - report, describing results of the whole pipeline
- Bowtie2-build --version 2.3.0 -building reference genome
- Bowtie2 --version 2.3.0 - mapping reads to reference genome
- Samtools --version 1.3.1 - manipulating alignments in the SAM files
- Bedtools --version 2.25.0 - enables genome arithmetic
- Igvtools --version 2.3.75 - preprocessing the data and visualization
- MACS2 --version 2.1.1.20160309 - calling peaks
- DAStk - differential ATAC-Seq analysis
FastQC gives general quality metrics about your reads. It provides information about the quality score distribution across your reads, the per base sequence content (%T/A/G/C). You get information about adapter contamination and other overrepresented sequences.
For further reading and documentation see the FastQC help.
Output directory: results/fastqc
sample_fastqc.html- FastQC report, containing quality metrics for your untrimmed raw fastq files
zips/sample_fastqc.zip- zip file containing the FastQC report, tab-delimited data file and plot images
MultiQC is a visualisation tool that generates a single HTML report summarising all samples in your project. Most of the pipeline QC results are visualised in the report and further statistics are available in within the report data directory.
Output directory: results/multiqc
Project_multiqc_report.html- MultiQC report - a standalone HTML file that can be viewed in your web browser
Project_multiqc_data/- Directory containing parsed statistics from the different tools used in the pipeline
For more information about how to use MultiQC reports, see http://multiqc.info
TrimGalore is used for removal of adapter contamination and trimming of low quality regions. TrimGalore uses Cutadapt for adapter trimming and runs FastQC after it finishes.
MultiQC reports the percentage of bases removed by TrimGalore in the General Statistics table, along with a line plot showing where reads were trimmed.
Output directory: results/trimgalore
Contains FastQ files with quality and adapter trimmed reads for each sample, along with a log file describing the trimming.
sample_val_1.fq.gz,sample_val_2.fq.gz- Trimmed FastQ data, reads 1 and 2.
sample_val_1.fastq.gz_trimming_report.txt- Trimming report (describes which parameters that were used)
sample_val_1_fastqc.htmlsample_val_1_fastqc.zip- FastQC report for trimmed reads
Single-end data will have slightly different file names and only one FastQ file per sample:
sample_trimmed.fq.gz- Trimmed FastQ data
sample.fastq.gz_trimming_report.txt- Trimming report (describes which parameters that were used)
sample_trimmed_fastqc.htmlsample_trimmed_fastqc.zip- FastQC report for trimmed reads
bowtie2 is used to produce raw bam files, followed by various filtering steps (mappability and quality) to produce filtered bams.
Output directory: results/bowtie2
sample.sam- Alignment sam file
SAMtools is used for sorting and indexing the output BAM files from Bowtie2. In addition, the numbers of features are counted with the idxstats option.
Output directory: results/samtools
sample.sorted.bam- Sorted bam file
sample.sorted.bam.flagstat- Flagstat of the bam file
bedtools is used to generate BedGraph copies for the downstream analysis.
Output directroy: results/bedtools
sample.sorted.bed- BedGraph copies (easier for data analysis)
igvtools toTDF converts a sorted data input file to a binary tiled data (.tdf) file.
Output directory: results/igvtools
sample.tdf- binary tiled tdf data file
macs2 is a program for detecting regions of genomic enrichment. Though designed for ChIP-seq, it works just as well on ATAC-seq and other genome-wide enrichment assays that have narrow peaks. The main program in MACS2 is callpeak, and its options are described below. As input, MACS2 takes the alignment files produced in the previous steps. However, it is important to remember that the read alignments indicate only a portion of the DNA fragments generated by the ATAC. Therefore, we must consider how we want MACS2 to interpret the alignments.
Output directory: results/macs2
sample_peaks.xls- Tabular file which contains information about called peaks. Information include:
- chromosome name
- start position of peak
- end position of peak
- length of peak region
- absolute peak summit position
- pileup height at peak summit, -log10(pvalue) for the peak summit (e.g. pvalue =1e-10, then this value should be 10)
- fold enrichment for this peak summit against random Poisson distribution with local lambda, -log10(qvalue) at peak summit
- Tabular file which contains information about called peaks. Information include:
sample_peaks.narrowPeak- BED6+4 format file which contains the peak locations together with peak summit, pvalue and qvalue.
sample_summits.bed- BED format file which contains the peak summits locations for every peaks.
sample_peaks.broadPeak- BED6+3 format file which is similar to narrowPeak file, except for missing the column for annotating peak summits.
sample_peaks.gappedPeak- BED12+3 format file which contains both the broad region and narrow peaks.
sample_model.r- R script with which a PDF image about the model based on your data can be produced.
.bdg- bedGraph format files which can be imported to UCSC genome browser or be converted into even smaller bigWig files.
Refer to https://github.com/taoliu/MACS for the specifications of the output fields.
DAStk is a differential ATAC-seq toolkit, can be used to identify changes in TF activity across differential ATAC-seq datasets.
Output directory: results/md_scores
sample_Treatment_md_scores.txt- MD-scores of the differential analysis on ATAC-seq datasets
MA plotthat labels the most significant TF activity changes, at a p-value cutoff of 1e-7. Note that the condition names (DMSO and Treatment) were the same ones used earlier as the second half of the prefix.

barcode plotof each of these statistically significat motif differences that depicts how close the ATAC-seq peak centers were to the motif centers, within a 1500 base-pair radius of the motif center.
